
Эпидемиологические исследования убедительно показали, что курение является одним из главных факторов риска развития атеросклероза и тромбоза [1—3]. Несмотря на многочисленные исследования, точный механизм влияния табачного дыма на развитие атеросклероза остается не раскрытым.
Табачный дым содержит около 4000 различных химических веществ [4]. Наибольшую их часть составляет никотин (0,7—4,8%, в зависимости от марки сигарет), монооксид углерода CO (3—6%), диоксид углерода CO2 (10—15%), цианистый водород (0,1—0,2%), смолы — бензапирен, бензен (4,8—65,6 мг на сигарету) [5], радиоактивный полоний, свинец, висмут [1]. В процессе курения формируются свободные радикалы [4]. Какой вклад в развитие атеросклероза вносит каждое из этих веществ, до сих пор неясно. Установлено, что свободные радикалы и бензапирен являются наиболее вредными для дыхательных путей [6]. CO и CO2 присоединяются к гемоглобину и ухудшают обеспечение тканей достаточным количеством кислорода. Кроме того, CO повышает концентрацию некоторых белков в крови (фибриногена, a1-антитрипсина, гаптоглобина, церулоплазмина), что способствует воспалительным реакциям [7]. Из-за воздействия никотина в бо`льшем количестве формируются свободные радикалы, повреждающие митохондриальную ДНК [8, 9] и окисляющие липиды [10—12].
Никотин также действует на нервную систему. Повышая количество допамина в головном мозге, он увеличивает концентрацию адренокортикотропина, кортизола [13], норадреналина [14] и вазопрессина [15] в крови, приводя к сужению коронарных артерий и повышению артериального давления. В процессе атерогенеза важным является воздействие никотина и смол на клетки, находящиеся в контакте с кровью. Никотин активирует тромбоциты [16—20], изменяет количество разных веществ, синтезируемых в клетках эндотелия, в макрофагах, моноцитах [21, 22] и влияет на активность некоторых ферментов [23].
Перечисленные пути воздействия активируют два основных процесса: пролиферативно-воспалительную реакцию на повреждение сосудистой стенки и отложение в ней липидов. Воздействие никотина и смол на эти процессы исследовано лучше других. Цель настоящей статьи — на основе результатов последних исследований обобщить представления о влиянии никотина и смол на развитие атеросклероза.
Направления воздействий никотина и смол
Воздействие никотина и смол на развитие атеросклероза можно разделить на 3 основные группы (рис. 1). Надо отметить, что очень важной мишенью всех воздействий являются тромбоциты, эндотелиоциты, гладкие мышечные клетки (ГМК) кровеносных сосудов, макрофаги и моноциты. Эти структуры подвергаются воздействию никотина через никотиновые рецепторы ацетилхолина [16]. Механизмы влияния смол еще не установлены.

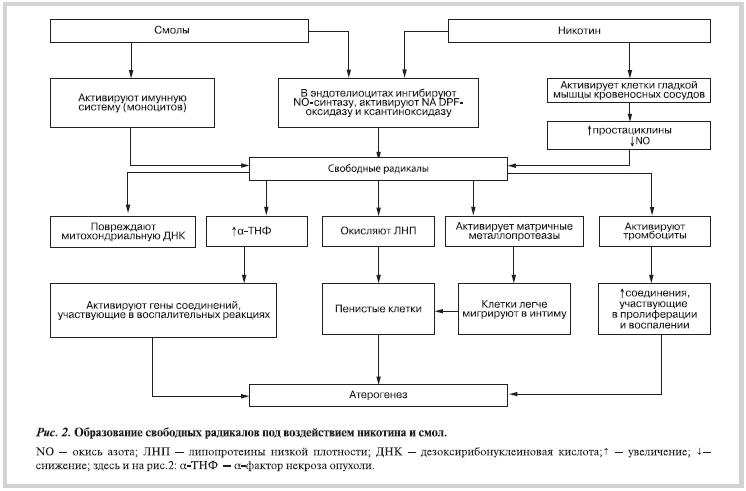
Образование свободных радикалов и их воздействие
Никотин и смолы побуждают образование свободных радикалов, влияя на активность ферментов или на экспрессию генов в тромбоцитах, эндотелиоцитах, ГМК кровеносных сосудов, макрофагах и моноцитах (рис. 2).
Никотин и смолы через разные промежуточные соединения ингибируют эндотелиальную NO-синтазу [23, 24], глутатионпероксидазу [25], активируют ксантиноксидазу [26, 27], липоксигеназу [28], в моноцитах и эндотелиоцитах — NADPH-оксидазу [29]. Оксид азота NO в эндотелиоцитах синтезируется NO-синтазой, коферментом которой является тетрагидробиоптерин [23, 30, 31]. Установлено, что никотин, уменьшая количество тетрагидробиоптерина, побуждает NO-синтазу продуцировать свободные радикалы [30], которые повреждают ДНК [2, 9, 32], а клетки начинают синтезировать больше сосудосужающих и провоспалительных веществ. Кроме того, смолы активируют клетки иммунной системы (моноциты), превращая их в источники свободных радикалов, которые повреждают эндотелиоциты [21], активируют тромбоциты [33], стимулируют пролиферацию ГМК кровеносных сосудов [34], а также окисляют липопротеиды низкой плотности (ЛНП) и ускоряют пероксидацию липидов [1]. Макрофаги, находящиеся под эндотелием, фагоцитируют окисленные ЛНП и становятся пенистыми (ксантомными) клетками (см. рис. 2). Из-за ухудшения синтеза NO, нарушения активности ферментов-антиоксидантов в эндотелиоцитах нарушается равновесие между окисленными и восстановленными соединениями. В этих клетках увеличивается количество пероксинитрита и нитротирозина, которые образуются вследствие воздействия NO с анионом супероксида; повышается количество маркеров, указывающих на перекисное окисление липидов — 8-изо-простагландина (8-изо-PGF2a) и окисление ДНК — 8-гидрокси-2-деоксигуанозина (8-OНdG) [21, 33]. Снижение синтеза NO в эндотелиоцитах приводит к снижению отношения между восстановленными и окисленными формами глутатиона в тромбоцитах, что вызывает окислительный стресс в этих клетках [19]. Тромбоциты активируются, выделяют соединения, участвующие в пролиферации ГМК кровеносных сосудов (тканевый фактор), в воспалительных реакциях (цитокины), в адгезии и агрегации тромбоцитов (тромбоксан А2 —TxA2, фибриноген) а также в свертывании крови (факторы свертывания тромбоцитов) [35].
Важно отметить, что на поверхности тромбоцитов расположены рецепторы для липопротеинов высокой плотности. Эти рецепторы необходимы для поддержания нормальной функции тромбоцитов и препятствуют образованию тромбов [36]. Предполагается, что свободные радикалы могут повредить эти рецепторы и окислять полиненасыщенные жирные кислоты, содержащиеся в мембранах клеток, нарушая таким образом деятельность клеток [10, 36].
Свободные радикалы способствуют синтезу ядерного фактора каппа-бетта (NF-κβ), который активирует многие гены, кодирующие соединения воспалительных реакций, например ген IL-8 [37]. Влияние свободных радикалов на экспрессию генов, продукты которых важны в процессе атерогенеза, не зависит от причины их появления. Никотин также влияет на экспрессию генов, продукты которых важны для транспорта электронов в дыхательной цепи митохондрий. В результате этого воздействия повышается внутриклеточное количество свободных радикалов [38].
Обобщая изложенное, можно утверждать, что никотин и смолы участвуют в образовании свободных радикалов, подавляя NO-синтазу и действуя на активность ферментов антиоксидантной системы (подавляя глутатионпероксидазу, активируя NADPH-оксидазу, ксантиноксидазу и липооксигеназу). Кроме того, смолы активируют моноциты, а никотин влияет на экспрессию генов, важных для транспорта электронов в митохондриях.
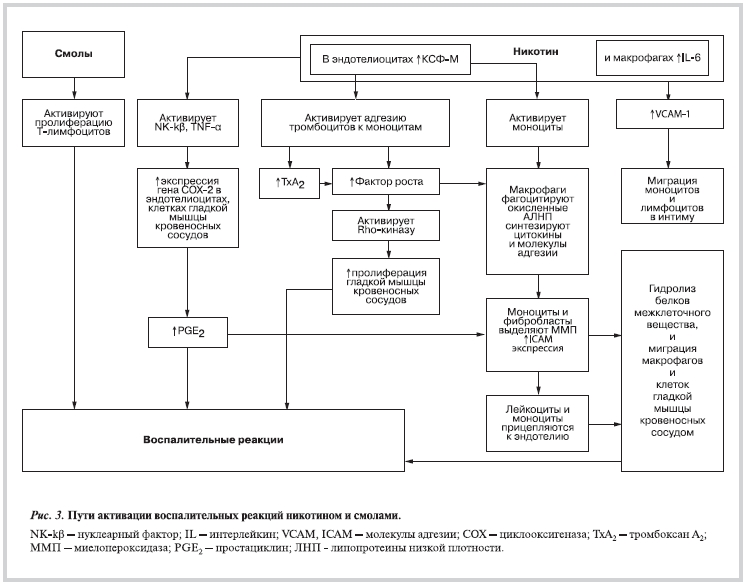
Bозбуждение воспалительных реакций
Никотин и смолы способствуют развитию воспалительных реакций через Т-лимфоциты, эндотелиоциты, макрофаги, ГМК кровеносных сосудов [22, 39, 40] (рис. 3).
В результате воздействия никотина, эндотелиоциты, макрофаги и клетки альвеолярного эпителия выделяют больше интерлейкина-6 (IL-6), колониестимулирующего фактора макрофагов (КСФ-M) [41—44], NK-κβ и α-фактора некроза опухоли (TNF-a) [45—47]. IL-6 является маркером воспалительного процесса [41, 44]. Он активирует синтез белков острой фазы в печени [48], а в эндотелиоцитах — синтез молекул адгезии (например, молекулы адгезии кровеносных сосудов — VCAM-1) [49], которые принимают участие в миграции моноцитов и лимфоцитов в интиму [48, 50]. КСФ-М активирует моноциты, стимулирует адгезию тромбоцитов к моноцитам, поэтому тромбоциты начинают выделять TxA2
[33]. КСФ-М оказывает локальное и системное действие. Локально КСФ-М способствует дополнительной секреции TxA2 [33] и дифференцированию моноцитов в макрофаги [51]. Систематическое воздействие КСФ-М заключается в повышении способности макрофагов фагоцитировать окисленные ЛНП [42]. Все эти факторы играют важную роль в процессе атерогенеза.
Из-за нарушения окислительно-восстановительных процессов в клетках иммунной системы инициируются воспалительные процессы в сосудистой стенке. В ответ на повышенное количество свободных радикалов в крови увеличивается количество окисленных ЛНП и маркеров воспаления [11]. Моноциты и лимфоциты начинают синтезировать цитокины [48], а эндотелиоциты — молекулы адгезии [48, 46]. Эти вещества участвуют в регуляции активности эндотелиоцитов, в адгезии моноцитов и их миграции под эндотелием [19, 52]. Моноциты и фибробласты, подвергшиеся воздействию цитокинов и фактора роста, выделяют матричные металлопротеазы [23, 48]. Эти ферменты гидролизируют белки межклеточного вещества, что способствует миграции ГМК сосудов и макрофагов [10, 53]. Выяснено, что матричные металлопротеазы освобождаются и под прямым воздействием никотина [54, 55]. Кроме того, в крови курящих людей уменьшается концентрация витамина С, с чем связано снижение синтеза коллагена-1 [55].
Смолы усиливают экспрессию митохондриальной ДНК, приводя к увеличению роста культуры клеток и размножения под воздействием митогенов [8]. Важно отметить, что соединения, находящиеся в табачном дыме, активируют пролиферацию ГМК сосудов и через фермент Rho-киназу, т.е., через ферментную систему, которая регулирует множество сигнальных путей, контролирующих рост, воспроизведение и другие функции клеток [56]. Rho-киназу активируют тромбоцитарный фактор роста, выделяемый активированными тромбоцитами [57], и 8-iso-PGF2a [33]. Из-за воздействия никотина на тромбоциты эти клетки начинают выделять больше фактора роста. Он способствует миграции ГМК сосудов в интиму [58].
На поверхности эндотелиоцитов и лейкоцитов никотин повышает количество межклеточных молекул адгезии (ICAM) и VCAM-1 [52, 59] , что также способствует миграции моноцитов и лимфоцитов под эндотелием [48]. Экспрессия VCAM-1 увеличивается из-за повреждения эндотелия, который меньше синтезирует и выделяет NO [30, 45]. В зоне промотора гена VCAM-1 находятся несколько областей для присоединения факторов транскрипции, в том числе NF-κβ [50]. Известно, что NO ингибирует NF-κβ, поэтому экспрессия гена VCAM-1 подавляется [50, 59].
Никотин повышает экспрессию гена, продукт которого — фермент циклооксигеназа-2 (СОХ-2) — в разных клетках (эндотелиоцитах, моноцитах, макрофагах, ГМК кровеносных сосудов и др.) участвует в синтезе простагландинов [45]. В развитии воспалительных реакций важную роль играет влияние TxA2 и простациклина E2 (PGE2). PGE2 активирует экспрессию матриксных металлопротеиназ и ICAM [60]. Поэтому лейкоциты и моноциты прикрепляются к эндотелиоцитам, что способствует атерогенезу [52]. В зоне промотора гена COX-2 находится несколько областей для присоединения разных факторов, в том числе NF-κβ и IL-6 [45]. Никотин активирует экспрессию гена СОХ-2 через активацию NF-κβ [45]. Известно, что NF-κβ активирует синтез веществ, стимулирующих воспаление, таких как IL-6, α -ТNF и интерферон [47]. Кроме того, фермент СОХ-2 активируется NADPH-оксидазой, которую, в свою очередь, активируют свободные радикалы [29]. TxA2 активирует тромбоциты, стимулирует синтез тканевого фактора в фибробластах, и активирует ГМК кровеносных сосудов, вследствие чего дестабилизируются атеросклеротические бляшки [61].
Функции моноцитов и макрофагов регулируют простациклины [35]. В организме курильщиков большее количество PGH2 требуется для увеличенного синтеза TxA2 в агрегатах моноцитов с тромбоцитами [35], а это способствует синтезу фактора роста в фибробластах [61]. Таким образом, в артериальной стенке повышается число ГМК и количество синтезированного ими межклеточного вещества. В крови курильщиков также повышается уровень аденозиндифосфата (АДФ), выделяемого активированными тромбоцитами. Важно отметить, что повышенная концентрация АДФ способствует агрегации тромбоцитов [62].
Никотин, действуя через никотиновые рецепторы ацетилхолина, увеличивает число рецепторов P2Y12 на поверхности тромбоцитов, эндотелиоцитов и ГМК кровеносных сосудов [16]. Показано, что никотин связывается с никотиновыми рецепторами ацетилхолина на поверхности мегакариоцитов, из которых в костном мозге формируются тромбоциты, имеющие больше рецепторов P2Y12. Этот феномен наблюдается только в результате регулярного курения, так как только в таких случаях повреждаются мегакариоциты [18]. Увеличивая количество рецепторов P2Y12 на поверхности эндотелиоцитов и ГМК кровеносных сосудов, никотин активирует эти клетки [16]. Клетки ГМК кровеносных сосудов начинают размножаться (см. рис. 3); они синтезируют и выделяют больше коллагена, приводя к утолщению артериальной стенки. Активированные эндотелиоциты синтезируют и выделяют больше тканевого фактора эндотелина-1, активирующего тромбоциты [20], а макрофаги — IL-1, α -ТNF [11]. Эти вещества активируют тромбоциты, стимулируют местное воспаление, и следовательно, процессы атерогенеза. Недавно установлено, что в макрофагах и миофибробластах число сегментов рецептора, участвующих в присоединении никотина, в атеросклеротических зонах увеличивается [22].
Обобщая воздействие никотина и смол на клетки, участвующие в воспалительных реакциях, можно отметить, что они выделяют различные соединения (IL-6, TNF-a, NF-κβ, протеазы) и экспрессируют молекулы адгезии (ICAM, VCAM-1), которые и инициируют воспалительные реакции. Пролиферация ГМК сосудов в организме курильщиков осуществляется и через активацию Rho-киназы. Кроме того, никотин усиливает экспрессию генов VCAM-1 и СОХ-2, увеличивает число рецепторов P2Y12 на поверхности тромбоцитов, эндотелиоцитов и ГМК кровеносных сосудов, что способствует воспалительным реакциям в развитии атеросклероза.
Другие воздействия
Соединения, находящиеся в табачном дыме, также влияют на экспрессию других генов, продукты которых важны в процессе атерогенеза. Например, в мегакариоцитах курящих людей активируются ферменты, деметилирующие промотор гена моноаминооксидазы (МАО) [63], т.е., курение активирует экспрессию этого гена. МАО инактивирует биологически активный амин серотонин, участвующий в воспалительных реакциях организма, превращая его в 5-гидроксииндолуксусную кислоту [63]. В тромбоцитах, произошедших из мегакариоцитов курильщиков, серотонин инактивируется быстрее. Такое изменение метаболизма серотонина в тромбоцитах можно объяснить как защитную реакцию организма против воспаления, инициированного соединениями, находящимися в табачном дыме [64].
Никотин ухудшает и комуникационные возможности между эндотелиоцитами, так как уменьшает экспрессию конексинов Сх 37 и Сх 43 на их поверхности [65].
Заключение
Никотин и смолы стимулируют атерогенез, усиливая формирование свободных радикалов и способствуя развитию воспалительных реакций. Никотин, действуя через никотиновые рецепторы ацетилхолина, активирует тромбоциты, эндотелиоциты и гладкие мышечные клетки кровеносных сосудов, вследствие чего выделяется большее количество соединений, способствующих воспалению, свертыванию крови и пролиферации гладких мышечных клеток сосудов (тромбоциты выделяют больше тромбоксана A2, фактора роста, эндотелиоциты — простациклинов, эндотелина-1, активирующего фактора тромбоцитов, гладкие мышечные клетки — коллагена). Никотин также активирует макрофаги, альвеолярные эпителиальные клетки, которые выделяют больше соединений, способствующих воспалению и фагоцитозу (интерлейкин-6 и колониестимулирующий фактор макрофагов). Смолы ингибируют NO-синтазу, активируют NADPH-оксидазу и ксантиноксидазу, поэтому усиливается перекисное окисление липидов и активируются матриксные металлопротеиназы, из-за чего гладкие мышечные клетки легче мигрируют. На основе этих исследований составлены схемы (см. рис. 2, 3), объединяющие воздействия никотина и смол на атерогенез. Новейшие научные данные подтвердили, что соединения, находящиеся в табачном дыме, способствуют атерогенезу и через воздействие на экспрессию генов (Rho-киназы, МАО, VCAM-1, ICAM и СОХ-2).



{kind=link}
{kind=link}