
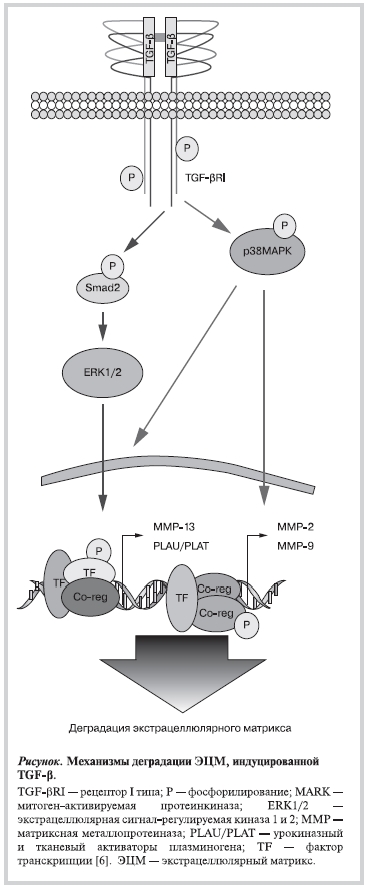
Семейство трансформирующего β-фактора роста (TGF-β) включает группу гомологичных гетеродимерных белков TGF-β1, -β2 и -β3, которые были так исходно названы по их способности активировать рост фибробластов, стимулируя трансформацию клеток [1]. Эти белки экспрессируются в миофибробластах, гладких мышечных клетках сосудов, эндотелиальных клетках и макрофагах. Влияние TGF-β на клетку обеспечивается благодаря сигнальному пути TGF-β, включающему специфичные мембранные рецепторы и внутриклеточные медиаторы и регулирующему множество биологических процессов, в том числе морфогенез, эмбриональное развитие, дифференцировку стволовых клеток, иммунный ответ, воспаление, а также формирование и деградацию экстрацеллюлярного матрикса (ЭЦМ). Рецепторами к TGF-β являются мембранные серин-/треонинкиназы I и II типов (TBRI и TBRII соответственно), а внутриклеточный этап сигнального пути TGF-β подразделяется на классический и альтернативный. Классический путь включает активирование белков Smad2 и Smad3, их гетеромеризацию с участием кооперирующего Smad4 и проникновение гетеромерного комплекса внутрь ядра, где они выполняют функцию активатора транскрипции генов, ответственных за формирование и деградацию ЭЦМ (табл. 1) [2, 3]. Однако TGF-β стимулирует деградацию ЭЦМ в первую очередь через не-Smad альтернативные пути, что приводит к повышенной экспрессии генов матричных металлопротеиназ (ММП), активатора плазминогена и др., вызывая протеолитическую деструкцию ЭЦМ (см. рисунок) [4—6]. Наличие многих уровней и различных механизмов регулирования формирования и деградации ЭЦМ делают сигнальный путь TGF-β (классический и альтернативные) ключевым звеном, влияющим на его строение.
Таблица 1. Влияние классического сигнального пути TGF-βна формирование ЭЦМ [6]

Примечание. TGF-β — трансформирующий β-фактор роста; ЭЦМ — экстрацеллюлярный матрикс; PAI-1 — ингибитор активатора плазминогена-1; TIMP — тканевый ингибитор металлопротеиназ; ММП — матричные металлопротеиназы.
Изменение активности различных компонентов сигнального пути TGF-β связано с целым рядом наследственных нарушений соединительной ткани. При синдроме Марфана из-за нарушения функции фибриллина, обусловленного различными мутациями гена FBN-1, нарушается секвестрация латентного комплекса TGF-β, что делает его более доступным для активации и ведет к повышению его концентрации. Считается, что целый ряд проявлений синдрома Марфана, в частности аневризма аорты, обусловлены именно повышением активности сигнального пути TGF-β [7]. Синдром Льюиса—Дитца 1-го и 2-го типа, выделенный недавно из синдрома Марфана, вызван мутациями в обеих субъединицах рецептора к TGF-β (TBR1 и TBR2 соответственно). И хотя мутантные рецепторы TGF-β не в состоянии передавать сигналы внутрь клетки, неизмененные рецепторы (они также экспрессируются у гетерозиготных пациентов) чрезмерно активно реагируют на TGF-β, что фактически выражается в повышенной активности сигнального пути TGF-β [8]. Миксоматозные изменения в митральном клапане (МК), приводящие к его недостаточности, при этом наблюдаются преимущественно при мутации гена FBN-1, но не TBR2 [9]. Повышение концентрации TGF-β также наблюдается при сосудистом типе синдрома Элерса—Данло, что может быть обусловлено попыткой компенсировать отсутствие коллагена III типа у этих пациентов [6].
Влияние сигнального пути TGF-β на формирование пролапса МК (ПМК) изучено мало. Имеются данные о его активности в миксоматозно измененных створках при синдроме Марфана, на FBN-1C1039G/+ и FBN-1C1039G/C1039G мутантных мышах [10]. Сходные данные получены при исследовании активности внутриклеточного сигнального пути TGF-β в створках МК, участки которых были резецированы в ходе реконструктивной операции при первичном ПМК. По данным S. Rizzo и соавт., при исследовании 15 миксоматозных МК выявлены высокая плотность окрашивания ядра по фосфорилированной активной форме Smad2 (38% по сравнению с контролем — 12%; p<0,0001) и нарушенная экспрессия TGF-β-связанных генов (по данным полимеразной цепной реакции) в миофибробластах, что свидетельствует о повышенной активации сигнального пути TGF-β в клетках миксоматозно измененных створок [11]. В миксоматозных створках, по данным K. Obayashi и соавт., выявляется также повышенная экспрессия TGF-β3 и TВR-II [12]. Кроме того, изменения МК могут наблюдаться и при нарушении функционирования внутриклеточных медиаторов сигнального пути TGF-β. По данным K.M. Glavin и соавт,. у мышей с недостатком ингибиторного Smad6 наблюдается гиперплазия МК [13]. В то же время в исследовании H.T. Chou и соавт. не выявлено связи между ПМК и полиморфизмом гена TGF-β1 (C-509T и T869C) в китайской популяции [14].
В отечественной литературе также имеется ряд указаний на повышение концентрации TGF-β в сыворотке крови при ПМК. Так, в работе А.В. Ягода и соавт. показано достоверное повышение уровня TGF-β1 у 220 молодых пациентов с ПМК (4,4±0,5 нг/мл) по сравнению с контролем (0,5±0,2 нг/мл; р<0,05) [15].
Кроме того, TGF-β является профибротическим цитокином, который стимулирует продукцию белков ЭЦМ в различных органах и системах, чрезмерная экспрессия которого приводит к развитию фиброза тканей [16]. Так, повышенная экспрессия TGF-β1 помимо утолщения створок и их дисфункции [17] вызывает формирование кардиального фиброза [18, 19], а увеличение содержания белков ЭЦМ в миокарде — появление его систолической и диастолической дисфункции [20].
В настоящее время ПМК служит наиболее частой причиной тяжелой неишемической митральной недостаточности (МН) и основной причиной хирургического вмешательства на МК из-за МН в Западной Европе и США [21]. Однако необходимо понимать, что реконструктивные операции, которые являются наиболее предпочтительными у пациентов с ПМК и тяжелой МН, не останавливают процесс миксоматозной дегенерации створок и что повышенная активность сигнального пути TGF-β может оказывать неблагоприятное воздействие на функцию левого желудочка (ЛЖ) даже после устранения его гемодинамической перегрузки.
Целью нашего исследования стали определение содержания растворимых активных форм TGF-β1 и TGF-β2 в сыворотке крови пациентов, прооперированных по поводу ПМК, осложненного тяжелой МН, и оценка взаимосвязи между концентрацией TGF-β1/2 и прогрессированием миксоматозных изменений и ремоделированием камер сердца после хирургического вмешательства.
Материал и методы
Обследованы 35 пациентов, наблюдаемых в консультативно-диагностическом центре ФЦСКЭ им. В.А. Алмазова после хирургической коррекции тяжелой МН, обусловленной первичным ПМК, не удовлетворяющие критериям синдрома Марфана и других наследственных нарушений соединительной ткани. Большинству пациентов (n=29) выполнена реконструктивная операция — имплантация опорного кольца Карпентье, у 6 она дополнена пластикой задней створки МК. Сроки обследования после операции колебались от 4 до 15 мес (средний 6,1±3,5 мес). Средний возраст обследованных составил 62,5±7,9 года, 46% мужчины.
Дополнительная группа сформирована из детей прооперированных пациентов. В эту группу вошли 11 человек (32,5±11,3 года, 64% мужчины), потомков первого поколения 10 прооперированных пациентов.
Всем обследованным выполняли эхокардиографию в 2D-, допплеровском и тканевом режимах (Vivid 7 Dimension, матричный фазированный датчик 3,5 МГц). Степень митральной регургитации (МР) оценивали в соответствии с рекомендациями Европейской эхокардиографической ассоциации по оценке клапанной недостаточности [22]. Размеры и объемы камер сердца, а также фракцию выброса ЛЖ измеряли в соответствии с рекомендациями Американского эхокардиографического общества по измерению камер сердца [23]. ПМК диагностировался при максимальном систолическом смещении створок за линию кольца более чем на 2 мм в парастернальном продольном сечении. Толщину створок МК измеряли в диастолу, в их средней части, вне зоны отхождения хорд, создающих ложное впечатление об их утолщении. Оценку деформации миокарда проводили с помощью методики spackle tracking при частоте кадров серошкального изображения 50—55 в секунду на рабочей станции EchoPAC’08.
Содержание TGF-β1 и TGF-β2 в сыворотке крови определяли иммуноферментным методом, используя тест-системы Human TGF-β1 Platinum ELISA и Human TGF-β2 Platinum ELISA. Исследования выполняли на автоматическом иммуноферментном анализаторе ELx 800.
Данные представлены как среднее ± стандартное отклонение, достоверность различий между ними оценивали с помощью критерия Стьюдента. Значимость различий между группами по качественным признакам определяли при помощи непараметрических методов (критерий Манна—Уитни). Различия между группами по частоте изучаемого признака определяли по критерию χ2. Линейную взаимосвязь двух количественных переменных оценивали с помощью коэффициента корреляции Пирсона, качественных переменных — с помощью коэффициента ранговой корреляции Спирмена. Статистическая обработка данных проведена с использованием программы Statistica 8.0.
Результаты
В группе пациентов, прооперированных по поводу ПМК, повышение концентрации TGF-β было выявлено у 23 (65%). Нормальные уровни TGF-β1 и TGF-β2 определялись лишь у 12 (35%) пациентов данной группы (см. табл. 2). Концентрация обеих изоформ TGF-β оказалась взаимосвязанной с морфологией МК после операции. Так, повышение концентрации TGF-β1 достоверно коррелировало с толщиной задней створки МК (r=0,67; p=0,016), а концентрация TGF-β2 — с резидуальным (сохраняющимся после реконструктивной операции) прогибом створок МК (r=0,68; p=0,007). Уровень TGF-β2 также умеренно положительно коррелировал с величиной резидуальной МР (r=0,56; p=0,01), а уровень TGF-β1 и TGF-β2 — с размером левого предсердия (r=0,48; p=0,03 и r=0,55; p=0,01 соответственно).
Таблица 2. Концентрация в сыворотке крови и доля повышенных уровней обеих изоформ TGF-β в обследованных группах
Примечание. Здесь и в табл. 3 ПМК — пролапс митрального клапана; * — различия между обследованными группами достоверны.
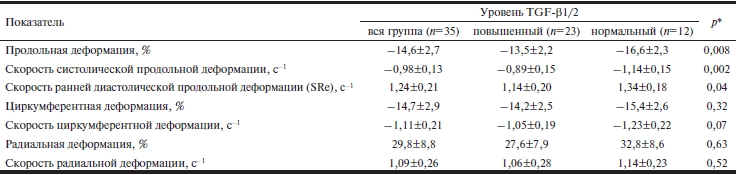
Повышение концентрации хотя бы одной из изоформ TGF-β отрицательно коррелировало с ударным объемом ЛЖ (r=–0,47; p=0,04). У лиц с нормальными уровнями TGF-β он был достоверно больше, чем у лиц с повышением концентрации одного из изучаемых цитокинов (72,6±15,9 и 57,0±14,1 мл соответственно; р=0,03). Множественный регрессионный анализ также выявил влияние концентрации TGF-β1 на фракцию выброса ЛЖ (β=0,93; р=0,04). Для более тонкого количественного анализа функции ЛЖ мы проанализировали показатели деформации миокарда ЛЖ. В табл. 3 представлены показатели глобальной систолической и диастолической продольной, радиальной и циркумферентной деформации и СД миокарда ЛЖ во всей группе и в подгруппах с нормальным или повышенным уровнем TGF-β1/2. Согласно приведенным данным в подгруппе пациентов с высоким содержанием TGF-β1 и/или TGF-β2 в сыворотке крови наблюдалось достоверное снижение продольной систолической и диастолической деформации и СД миокарда по сравнению с таковыми у пациентов с нормальным уровнем TGF-β1/2. Данные изменения являются точным количественным отражением ухудшения систолической и диастолической функции ЛЖ, более чувствительным, чем традиционные эхокардиографические показатели, такие как фракция выброса ЛЖ, время изоволюметрического расслабления и даже тканевая допплерография [24]. Различий по радиальной и циркумферентной деформации и СД в зависимости от содержания TGF-β1 и/или TGF-β2 в сыворотке крови не выявлено.
Таблица 3. Показатели деформации миокарда у пациентов с ПМК после реконструктивной операции на МК
Примечание. * — различия между подгруппами с нормальным и повышенным уровнем TGF-β1/2 в сыворотке крови достоверны.
Уровень TGF-β2 в сыворотке крови также оказался связан с ремоделированием правых камер сердца. Выявлены положительные корреляции с размером правого предсердия (r=0,69; p=0,001), толщиной стенки правого желудочка (r=0,55; p=0,017) и тяжестью трикуспидальной регургитации (r=0,62; p=0,003). В группе пациентов с повышенным уровнем TGF-β1/2 чаще выявлялась диастолическая дисфункция правого желудочка (χ2=4,0; p=0,04).
Нами также выполнена оценка системных эффектов TGF-β у пациентов, прооперированных по поводу ПМК. У пациентов с повышением содержания в сыворотке крови хотя бы одной из изоформ TGF-β ожидаемо выявлялся больший диаметр восходящей аорты (34,6±3,5 мм) по сравнению с таковым у лиц с нормальным уровнем TGF-β1 и TGF-β2 (31,1±4,8 мм; р=0,04). Концентрация TGF-β1 была выше у пациентов с пролапсом трикуспидального клапана (8,3±5,8 и 53,2±15,9 нг/мл; р=0,01), а повышение уровня TGF-β1 положительно коррелировало с наличием срединно-базальных ложных хорд (rs=0,55; р=0,02), папиллярных мышц (rs=0,57; р=0,01), а также с изменениями костной системы (долихостеномелии): размахом рук (r=0,46; p=0,04), длиной нижнего сегмента туловища (r=0,53; p=0,016), а TGF-β2 — с соотношением длины стопы и роста (r=0,46; p=0,04).
При обследовании потомков пациентов с ПМК выявлено повышение концентрации TGF-β1 и/или TGF-β2 в сыворотке крови у 73% обследованных, причем средняя концентрация TGF-β2, была недостоверно выше, чем в группе прооперированных пациентов (см. табл. 2). В 3 случаях наблюдалось повышение концентрации одной из изоформ TGF-β у потомка в отсутствие повышения ее у родителя и лишь в одном случае повышение уровня TGF-β не выявлялось у потомка при наличии его у прооперированного родителя. При этом лишь у 4 (36%) человек при эхокардиографии выявлен пролапс или прогиб (≤2 мм) МК. Однако и в данной группе концентрация TGF-β1 сильно коррелировала с толщиной задней створки (r=0,77; p=0,01), с утолщением (≥5 мм) одной из створок (r=0,68; p=0,021) и тяжестью МР (r=0,69; p=0,018).
Обсуждение
ПМК у выбранной нами для изучения группы пациентов является наиболее надежной моделью. Выраженность миксоматозных изменений в МК у этих пациентов привела к формированию тяжелой МН и обусловила необходимость хирургической коррекции порока. Изоформы TGF-β1 и TGF-β2, являясь первым звеном сигнального пути TGF-β, отражают его активность в целом у обследованных пациентов. Наиболее значимым результатом в данном исследовании, по нашему мнению, является повышение концентрации в сыворотке крови, по крайней мере, одной из изоформ TGF-β у большинства пациентов, что может свидетельствовать об участии сигнального пути TGF-β в формировании данной патологии. Подтверждением тому служит выявленная взаимосвязь между повышением концентрации TGF-β1 и толщиной задней створки МК, миксоматозные изменения которой наиболее часто выявляются при первичном ПМК, и тенденция к расширению аорты при повышении концентрации TGF-β1/2.
То же самое относится и к прогрессированию миксоматозных изменений после реконструктивного вмешательства на МК. Взаимосвязь между резидуальным прогибом МК, величиной резидуальной МР и TGF-β2 подтверждает данные литературы, согласно которым основной причиной повторного появления значимой МР после реконструктивной операции служит прогрессирование миксоматозных изменений (утолщение и пролабирование) в створках МК [25]. Поскольку ремоделирование левого предсердия в значительной степени связано с тяжестью МР, становится понятной положительная корреляция между уровнями обеих изоформ TGF-β и размером левого предсердия. Взаимосвязь ремоделирования правых камер и TGF-β2 может быть обусловлена повышенным давлением в легочной артерии, которое составило после операции 36,6±7,1 мм рт.ст. и также коррелировало с уровнем TGF-β2 (r=0,67; p=0,015). Согласно данным литературы, резидуальная легочная гипертензия чрезвычайно распространена у пациентов, перенесших реконструктивную операцию по поводу ПМК. Так, по данным A.B. Goldstone и соавт., она выявляется у 46% пациентов и является независимым предиктором послеоперационной смертности и заболеваемости независимо от исходного давления в легочной артерии [26]. Таким образом, TGF-β (преимущественно изоформа TGF-β2) не только влияет на прогрессирование миксоматозных изменений после реконструктивной операции, но и оказался связанным со всей цепью последующих гемодинамических изменений, характерных для порока МК.
Влияние повышенной концентрации TGF-β на систолическую и диастолическую функцию ЛЖ осуществляется, вероятно, через его профибротические свойства. Изменение деформации при фибротических изменениях в миокарде показано недавно для ряда генетических и аутоиммунных заболеваний, протекающих с поражением сердца [27, 28]. Методика spackle tracking, как показано ранее, является точным и хорошо воспроизводимым способом оценки локальной и глобальной систолической и диастолической функции ЛЖ в норме и при такой патологии, как клапанные пороки сердца и кардиомиопатии [24].
Результаты анализа группы потомков подтверждают выявленные нами взаимосвязи между повышением концентрации TGF-β и морфологией МК, прежде всего задней створки, изменения которой наиболее часто выявляются при первичном ПМК.
Однако следует отметить, что средние значения концентрации обоих цитокинов в обследованной группе имели достаточно большой разброс (см. табл. 2), именно поэтому нам не удалось определить пограничное значение TGF-β1/2, превышение которого приводит к патологическим изменениям створок МК. Существенным ограничением данного исследования также может быть небольшая численность обследованных групп пациентов.
Выводы
1. Трансформирующий β-фактор роста оказывает существенное влияние на прогрессирование миксоматозных изменений митрального клапана у пациентов, которым была выполнена реконструктивная операция по поводу тяжелой митральной регургитации при пролапсе митрального клапана.
2. Высокая активность сигнального пути трансформирующего β-фактора роста сочетается со снижением сократительной способности левого желудочка, что обусловлено, вероятно, его профибротической активностью.



{kind=link}
{kind=link}
{kind=link}