
С момента выхода в свет монографии «Mischqueschwulsteder Niere», которую Max Wilms опубликовал в 1899 г., наиболее частым сóлидным новообразованием мочевой системы у детей оставалась опухоль Вильмса (WT) – 3,9–10,9 случая на 1 млн детей [1, 2]. В современной структуре опухолевой патологии заболевание встречается в 9% наблюдений у детей до 1 года (4-е место) и лишь у 6% больных заболевание носит билатеральный характер [3]. В одном из последних аналитических исследований, проведенных Е. Ward et al. в 2014 г., показано, что пик встречаемости опухоли Вильмса приходится на возраст до 5 лет – 75% наблюдений [4]. В настоящее время разработаны протоколы комплексного ведения пациентов с WT (SIOP, NWTS). При выявлении заболевания на ранних этапах лечение успешно [5, 6]. Важным является то, что развитие данного онкологического заболевания ассоциируется с генетическими изменениями, а именно с мутацией гена, расположенного на коротком плече хромосомы 11 в 13-м локусе (позже были выявлены и другие мутации). Со временем были описаны случаи сочетания WT и других заболеваний, в совокупности обусловливающих более тяжелое течение болезни. Все они имели схожие генетические проявления – мутация гена WT1. В 1967 и 1970 гг. один из таких синдромов был описан двумя специалистами и в итоге был назван их именами – синдром Denys–Drash (DDS), который характеризуется триадой: нефропатией, нарушением формирования пола 46 XY и нефробластомой [7, 8].
Представляем клиническое наблюдение пациента с двусторонней метахронной опухолью Вильмса, нарушением формирования пола 46 XY в виде гипоспадии (мошоночная форма) и двустороннего абдоминального крипторхизма без нефропатии.
Пациент родился 1.12.2011, второй ребенок в семье (первый мальчик не имеет урологической патологии). После рождения у него была выявлена экстрагенитальная патология в виде двустороннего крипторхизма и гипоспадии (мошоночная форма). Ребенка готовили к плановому хирургическому лечению. Однако в возрасте 6 мес. во время купания ребенка мама обнаружила опухолевидное образование в животе. Проведено УЗИ, выявлена опухоль брюшной полости.
В дальнейшем ребенок госпитализирован в Детскую республиканскую клиническую больницу (ГАУЗ ДРКБ МЗ РТ). Была выполнена рентгеновская КТ (РКТ) брюшной полости и обнаружена онкоурологическая патология в сочетании с пороками развития мочеполовой системы (рис. 1). По результатам обследования поставлен диагноз «нефробластома слева T3NХM0. Двусторонняя брюшная ретенция яичек. Гипоспадия, мошоночная форма». 25.06.2012 ребенку в возрасте 7 мес. жизни проведена операция по удалению опухоли почки с одновременным низведением левого яичка в мошонку. Масса опухоли была равна 520 г, составив около 10% массы тела ребенка. Гистологическое заключение: смешанная нефробластома. Послеоперационный период протекал без осложнений, и было принято решение не проводить послеоперационной полихимиотерапии.
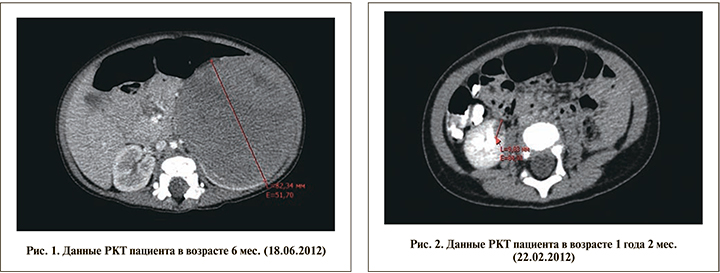
Через 8 мес. на контрольной РКТ брюшной полости в нижнем полюсе правой почки выявлен очаг округлой формы диаметром10 мм (рис. 2).
В связи с этим диагноз был скорректирован: «нефробластома двусторонняя метахронная T4NХM0. Двусторонняя брюшная ретенция яичек. Гипоспадия, мошоночная форма». 17.06.2013 проведена вторая операция: ретроперитонеоскопия с конверсией в мини-люмботомию, резекция нижнего полюса правой почки с опухолью, уретероуретероанастомоз с установкой мочеточникового стента. Через 2 мес., 14.08.2013, мочеточниковый стент был извлечен с использованием эндоскопической техники. Через год по результатам РКТ патологии не выявлено.
Затем проведено несколько пластических операций по формированию половых органов у ребенка. 15.07.2014 выполнено одноэтапное низведение яичка – правосторонняя орхопексия по Shoemaker при брюшной форме крипторхизма. А через 8 мес. начато формирование полового члена: осуществлена уретропластика Bracka-I, при которой использовался свободный кожный трансплантат крайней плоти. Послеоперационный период прошел без осложнений, трансплантат прижился. Спустя 10 мес. (18.01.2016) выполнен второй этап тубуляризирующей уретропластики.
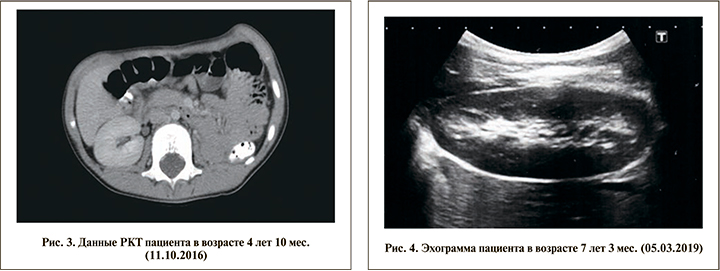
Ежегодно ребенок проходит диспансерное наблюдение в Республиканском центре детской урологии-андрологии на базе ГАУЗ ДРКБ МЗ РТ (рис. 3). По состоянию на март 2019 г. по результатам контрольного обследования (УЗИ) патологии брюшной полости не выявлено, отмечается викарная гипертрофия оставшейся части правой почки (рис. 4). Наружные половые органы сформированы по мужскому типу: половой член возрастных размеров, меатус открывается на головке полового члена, мочеиспускание свободное и безболезненное; гонады расположены в мошонке, размеры левого яичка соответствуют возрастным показателям, правое яичко гипоплазировано. Состояние мочевыделительной системы по данным биохимического анализа крови, анализов мочи оценено как удовлетворительное. Как и ранее, данных за протеинурию нет. Содержание мочевины и креатинина в пределах нормы.
В 2017 г. пациенту проведен поиск патогенных мутаций, ассоциированных с наследственными заболеваниями почек, а также с другими наследственными заболеваниями со сходными фенотипическими проявлениями. Анализ ДНК проведен методом парно-концевого чтения (2×125) со средним покрытием не менее 70–100×. Для пробоподготовки была использована методика селективного захвата участков ДНК, относящихся к кодирующим областям генов с известным клиническим значением.
Выявлена ранее не описанная гетерозиготная мутация в экзоне 7 гена WT1 (chr11:32417947G>A), приводящая к появлению сайта преждевременной терминации трансляции в 369-м кодоне (p.Arg369Ter, NM_024426.4). Гетерозиготные мутации в гене WT1, обусловливающие нарушение синтеза полноразмерного белка, описаны, в частности, у пациентов с DDS (OMIM: 194080) и WT, тип 1 (OMIM: 194070). Мутация не зарегистрирована в контрольных выборках «1000 геномов», ESP6500 и ExAC. Поскольку мутация нарушает синтез полноразмерного белка, ее следует расценивать как вероятно патогенную.
При DDS мутации гена WT1 чаще регистрируют в экзонах 8 и 9. Реже встречаются схожие по клиническим проявлениям с DDS мутации в других начальных экзонах локуса 11р13. Публикаций таких случаев немного, а классическая триада клинических проявлений неполная. К примеру, W. Bruening et al. [9] описали случай мутации в экзоне 7 у пациента женского рода c нефропатией, но без онкологии и нарушения формирования пола [9].
F. Auber et al. [10] представили несколько описаний клинических наблюдений, в том числе пациентов с мутацией в гене WT1 в неклассических экзонах 3, 4 и 7. У пациента с изменениями в экзоне 7 была выявлена точечная мутация, которая привела к замене аргинина 301 с формированием стоп-кодона при кариотипе 46 XY. Клинически заболевание проявлялось двусторонним паховым крипторхизмом (при этом в 19-месячном возрасте отмечалась гипоплазия яичек, в 16 лет – тестикулярная недостаточность), женским строением гениталий в виде влагалища (матка отсутствовала), двусторонней WT без нефротического синдрома (была выполнена односторонняя нефрэктомия и резекция контралатеральной почки) [10]. A. Takata et al. [11] в своей работе упоминают двух пациентов с мутацией в экзоне 7: один пациент мужского пола (46 XY) с манифестацией нефротического синдрома в возрасте 2 лет с быстрым прогрессированием до терминальной стадии, но без WT и нарушения формирования пола; второй пациент с похожей мутацией, с клиническими проявлениями нефропатии, нарушением формирования пола 46 XY, с женскими гениталиями без WT [11].
Наряду с вышеизложенным в литературе имеются другие описания мутаций гена WT1 в нехарактерных экзонах, но имеющих клинику, схожую с проявлениями DDS [12–15].
Представленные описания различных мутаций гена WT1, имеющих неклассические клинические проявления, позволяют объединить их в предложенный N. Bardeesy с коллегами термин «неполный DDS». В 1994 г. авторы использовали данный термин при описании пациентов с нефропатией, нарушением формирования пола или WT [11, 16]. Основным проявлением этого заболевания служит нефротический синдром как наиболее тяжелый фактор, не поддающийся медикаментозной терапии, устранить который можно только посредством пересадки почки.
В аналитических мультицентровых работах [9, 11, 12, 15] представлены случаи проявления нефропатии с нарушением формирования пола и/или WT. Во всех описаниях ведущим является нефротический синдром. Классический случай DDS был представлен J. Weaver et al. из детской больницы Вашингтона. У пациента с кариотипом 46 XY диагностированы дисгенезия гонад, терминальная стадия болезни почек и двусторонняя WT. С учетом генетически подтвержденного DDS (мутация в экзоне 8 в зоне 2 «цинкового пальца» гена WT1) выполнены билатеральная нефрэктомия и билатеральная гонадэктомия [17].
По сообщениям некоторых авторов, нефротический синдром может манифестировать позже или совсем не проявляться. Однако это возможно только при мутациях в нехарактерных экзонах. Так, специалисты из детской больницы Денвера (США) описали случай мутации в экзоне 6 гена WT1 у пациента с кариотипом 46 XY, неполным синдромом нечувствительности андрогенов, двусторонней WT (были проведены левосторонняя нефрэктомия и правосторонняя резекция почки) без нефротического синдрома [18]. Отсутствие нефропатии также было отмечено группой авторов Германии [19]. Из 53 пациентов, вошедших в исследование, у троих не было протеинурии. В двух случаях пациенты с кариотипом 46 XY имели мутации в экзоне 1 и двустороннюю WT. В одном случае пациент с нарушением формирования пола 46 XY с мутацией в экзоне 2 не имел онкологической патологии.
P. Dattolo et al. [20] описали мутацию в экзоне 6 с нестандартной клинической картиной. После выявления односторонней WT была выполнена нефрэктомия. Позже был обнаружен патологический процесс в единственной оставшейся почке, но данные гистологического исследования позволили исключить онкологическую патологию в пользу почечной дисплазии. Однако в возрасте 15 лет проявились симптомы нефропатии, которые со временем усилились. Потребовался гемодиализ, а позже была проведена пересадка почки. Случай демонстрирует нестандартные проявления DDS, даже в его неполных и перекрестных формах, и необходимость генетического исследования больных с WT и нефропатиями. В большинстве статей встречаются высказывания о нестандартных ситуациях в отношении пациентов с предположительной патологией гена WT1. В связи с чем коллектив авторов под руководством B. Köhler (2011) предлагают проводить генетические исследования всем пациентам с 46 XY дисгенензией гонад со структурными изменениями почек и/или протеинурией [21]. M. Finken et al. предлагают пересмотреть данное предложение и проводить генетические исследования пациентов с дисгенезией гонад даже если не выявлено патологии почек [22].
Таким образом, мутации в экзонах 8 и 9 всегда проявляются нефропатией, а мутации других участков WT1 и чаще всего вне зоны «цинковых пальцев» могут протекать без нарушений функции почек. В описанных выше случаях можно выделить отсутствие и/или позднее проявление нефропатии: имеется ли влияние нефрэктомии с одной стороны и резекции почки с противоположной на проявление и манифестацию нефротического синдрома? Вряд ли нефропатия может появиться в старшем возрасте, если она не закодирована генетически, но возможно влияние каких-либо факторов на сроки ее проявления. Это требует сопоставления большего количества случаев.
При рассмотрении вариаций DDS (полные/неполные/перекрестные случаи), причиной которых также служат мутации в гене WT1, некоторые авторы предлагают не разграничивать их, а также любые точечные мутации в этой зоне. Однако, поскольку данные синдромы различаются клинически и имеют разные подходы к лечению, вероятно, каждую из этих мутаций следует выделять в отдельную нозологию.
Как показал анализ литературы, представленное клиническое наблюдение не имеет ранее описанных аналогов, в настоящее время остается в статусе «de novo» и требует дальнейшего наблюдения. Для определения оптимальной тактики ведения пациентов с патологией гена WT1 необходимо сформировать международный архив данных редких мутаций.


