
Агенезия крестца (АК) – порок развития каудального отдела позвоночника и спинного мозга из группы аномалий, объединенных общим названием «синдром каудального регресса (СКР)». Кроме АК в данную группу аномалий входит агенезия копчика, а также вышерасположенных позвонков – поясничных и нижне-грудных [1–3]. Наиболее тяжелое проявление СКР сопровождается гипоплазией и сращением (костным или кожным) нижних конечностей и носит название «синдром русалки», или сиреномиелия [4]. АК – очень редкая аномалия с частотой встречаемости, по разным источникам, от 1 на 25 тыс. до 1 на 100 тыс. новорожденных [1, 5]. АК встречается как в изолированном варианте, так и сочетании с другими аномалиями (спинномозговыми грыжами, гидроцефалией, с пороками сердца, желудочно-кишечного тракта, мочевыделительной и половой систем). Наиболее характерно наличие дистальных атрезий при сиреномиелии (аноректальная атрезия, агенезия гонад, почек) [6]. Механизм развития указанных атрезий связывают с отсутствием или недостаточностью кровоснабжения каудальных структур – сбросом крови в плаценту через добавочный сосуд [1, 7]. Фактором риска появления данной аномалии является сахарный диабет у матери со стажем болезни более 10 лет, недостаточным контролем уровня гликемии с развитием ангиопатии до зачатия и тератогенным действием гипер-, гипогликемии и кетоновых тел на ранних сроках гестации с повреждением каудальной мезодермы [8]. Описан даже биохимический процесс повреждения при СКР, при котором происходит нарушение синтеза пиримидина в пентозофосфатном цикле [9–10]. Кроме того, к факторам риска относят употребление алкоголя беременной и прием ретиноевой кислоты [11]. Варианты АК изучены и впервые опубликованы T. Renshaw в 1978 г. Автор описал 4 типа агенезии: 1-й – односторонняя полная или частичная АК, 2-й – двусторонняя неполная АК, 3-й – полная АК и вариабельная поясничная агенезия с соединением подвздошных костей с последним имеющимся позвонком и 4-й тип – полная АК и вариабельная поясничная агенезия без соединения подвздошных костей с последним имеющимся позвонком. Исходя из представленного описания, при 1-м и 2-м типах – позвоночно-тазовое сочленение стабильно, при типе 3 – относительно стабильно, а при 4-м – нестабильно [12–14]. Данная классификация представляется наиболее удобной и понятной в применении. В дальнейшем предлагался ряд других классификаций. Так, A. Cama et al. в 1996 г. в дополнение к представленным вариантам отдельно выделил агенезию копчика [15], в 2002 г. J. Guille et al. соотнесли анатомический вариант аномалии с возможностью самостоятельного передвижения и разделили СКР на типы А, В и С, где при типе А пациенты передвигаются самостоятельно, при типе В такая возможность ограниченна, при типе С возможность передвижения полностью отсутствует [16]. Исходя из изложенного описания аномалии, очевидно, что у пациентов имеется неврологический дефицит нижних конечностей и тазовых органов. Степень выраженности нарушений соотносится с типом аномалии. Нарушение функции нижних конечностей проявляется от мышечной гипотонии до глубоких парезов и параличей. Выявляются эквиноварусные деформации стоп, сгибательные контрактуры коленных и тазобедренных суставов, врожденный вывих бедра, гипоплазия таза и нижних конечностей [17–18]. Основной задачей ортопедов при АК является вертикализация пациента с условием устранения нестабильности позвоночно-тазового соединения и коррекции деформаций нижних конечностей. Все не так однозначно с функцией детрузора. Публикации о наблюдении пациентов с АК обычно описывают за редким исключением один-два клинических наблюдения и, как правило, нейрохирурги ческую и ортопедическую составляющую проблемы. При поиске в различных библиотечных системах мы не нашли работ, указывающих на формирующиеся варианты нейрогенного мочевого пузыря, соответственно, и тактики ведения урологом детей с подобными аномалиями. В такой ситуации достаточно сложно составить какие-либо алгоритмы ведения данной категории пациентов урологом. В связи с этим считаем возможным поделиться с коллегами собственным клиническим наблюдением ребенка с АК с описанием нейроурологических нарушений.
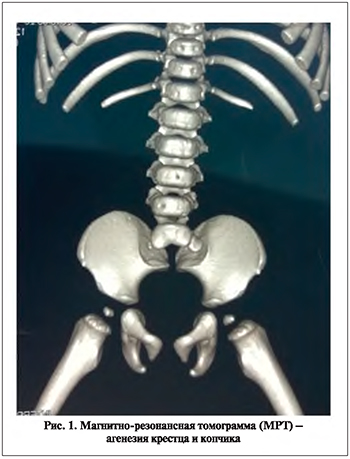
Девочка наблюдалась в возрасте с 1 года до 4,5 лет, проведено несколько госпитализаций в Детскую городскую клиническую больницу им. Сперанского Москвы и Детскую республиканскую больницу Петрозаводска. Представлено наблюдение АК 3-го типа по T. Renshaw (рис. 1). У ребенка не выявлены аномалии развития других органов и систем. К возрасту 1 года стало ясно, что самой существенной проблемой является нарушение функции тазовых органов, которое проявлялось постоянным выделением мочи каплями и наличием колостаза, который корригировали клизмами и механическим извлечением каловых масс из прямой кишки. На этом фоне с раннего возраста рецидивировала лейкоцитурия. При первичном обследовании в возрасте 1 года выявлен арефлекторно-дезадаптированный мочевой пузырь емкостью 50 мл с крайне высоким внутрипузырным давлением – до 214 см вод.ст. Позыва на мочеиспускание, эквивалента позыва не отмечено. Отсутствовали пузырно-мочеточниковые рефлюксы. Высокое внутрипузырное давление явилось показанием к назначению М-холиноблокаторов, но при попытке применения оксибутинина гидрохлорида отмечена выраженная атропиноподобная реакция, из-за которой продолжение лечения данным препаратом не представлялось возможным даже в минимальных дозах. Начата интермиттирующая катетеризация мочевого пузыря. Отношение к катетеризации ребенка было крайне негативным, назначенный режим катетеризаций родителями не соблюдался, и до 3-летнего возраста повторных госпитализаций в урологический стационар не было. Амбулаторно использованы различные методы физиолечения, уросептики, но динамики в функции мочевого пузыря не отмечено и практически постоянно выявлялась лейкоцитурия. В 3-летнем возрасте принято решение использовать М-холиноблокатор троспия хлорид. Подобрана доза и схема терапии, при которой не отмечено наличия побочных эффектов (2,5 мг 2 раза в день – 2 нед., затем – 1 мес. перерыв, после второго обследования перерыв уменьшен до 2 нед.). Применение троспия хлорида позволило улучшить функцию удержания мочи, увеличить емкость мочевого пузыря и снизить внутрипузырное давление. Кроме того, для того чтобы обеспечить самостоятельное мочеиспускание, в схему лечения включен препарат группы альфа-адреноблокаторов, в частности доксазозин в минимальной дозировке 0,16 мг (6 нед. – применение, затем – 2 мес. перерыв). На фоне применения данного препарата появился позыв на мочеиспускание. Указанные препараты в наших клиниках используются уже более 15 лет, они введены в формуляры больниц формулярными комиссиями, и при каждом назначении родители подписывают информированное согласие на применение данных препаратов. Представим описание уродинамического исследования в двух контрольных точках – через 6 и 12 мес. (ребенку 3,5 и 4 лет) и завершающее наблюдение исследования – через 1,5 года от начала лечения (в возрасте 4,5 лет).
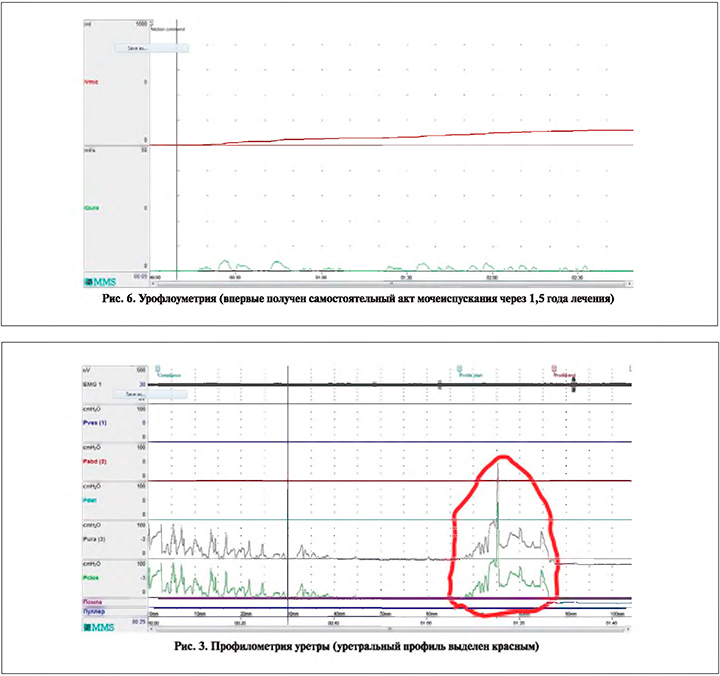
После первых 6 мес. лечения емкость мочевого пузыря составила 70 мл лежа и 75 мл сидя, восстановилась сенсорность мочевого пузыря. При цистометрии регистрировалось нормальное базовое внутрипузырное давление, на фоне которого выявлялись единичные незаторможенные сокращения детрузора с давлением до 20 см водн.ст. и предмикционным комплексом сокращений – проявления гиперктивности мочевого пузыря (рис. 2). При профилометрии уретры выявлялось нестабильное давление на протяжении профиля, но вполне удовлетворительное для замыкательной функции сфинктера (рис. 3). При этом самостоятельное мочеиспускание отсутствовало.
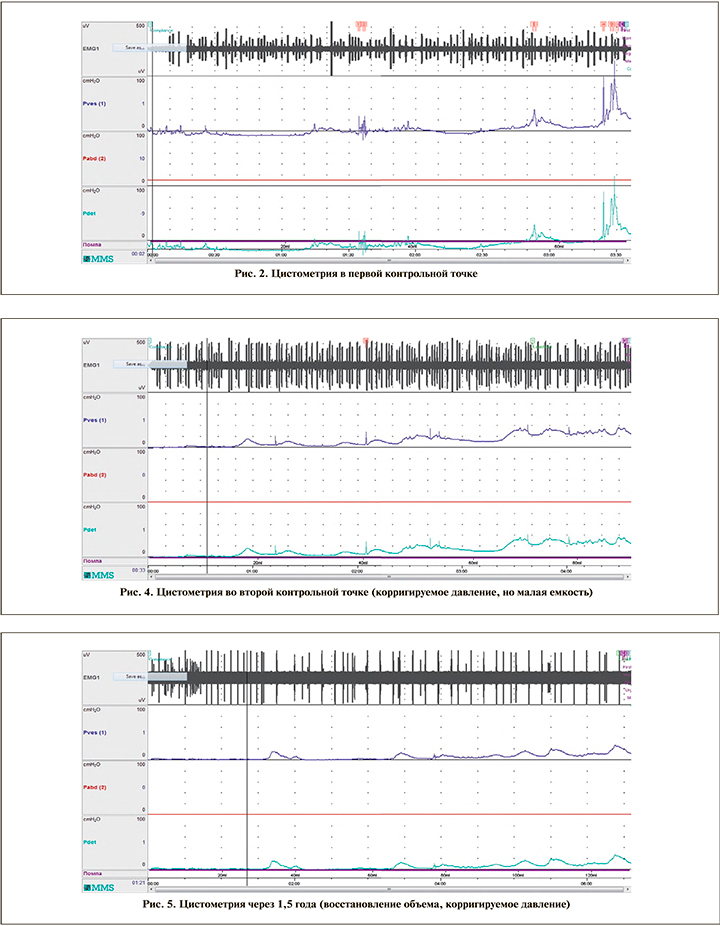
Через 12 мес. лечения емкость мочевого пузыря составила 80 мл. При нормальном базовом внутрипузырном давлении увеличилось количество незаторможенных сокращений (рис. 4), в связи с чем уменьшены перерывы в приеме троспия хлорида до 2 нед.
Через 1,5 года емкость мочевого пузыря составила 130 мл лежа и 139 мл сидя. Проявления гиперактивности мочевого пузыря уменьшились (рис. 5). При проведении урофлоуметрии впервые получен самостоятельный акт мочеиспускания – обструктивно-прерванный, но выделено 116 мл за 92 с (рис. 6). Количество остаточной мочи составило 20 мл. Таким образом, за 1,5 года выявлена выраженная положительная динамика функции детрузора.
Параллельно с применением М-холиноблокатора и альфа-адреноблокатора использованы курсы уросептиков, антибактериальных препаратов, слабительные средства. На фоне стабилизации уродинамики купирована лейкоцитурия, за последние 6 мес. наблюдения патологических изменений в анализах мочи не отмечено. Ультразвуковое исследование почек показало нормальный результат, не выявлено патологических изменений биохимического исследования крови.
Приводим основные предложенные рекомендации для амбулаторного этапа лечения:
- Режим питья — по 200–250 мл каждые 2 ч в дневное время, режим мочеиспусканий – после каждого приема жидкости.
- Дополнительно курсами – массаж живота, ног.
- Доксазозин на ночь 0,16 мг – 6 нед., затем перерыв 2 мес. и повторные 6-недельные курсы.
- Троспия хлорид по 2,5 мг 2 раза в день – 2 нед., далее 4-недельный перерыв, затем повторные курсы (условия применения доксазозина и троспия хлорида – см. выше – официальное разрешение формулярных комиссий и информированное согласие родителей).
- Использование уросептиков по ситуации.
- Контроль общего анализа мочи 1 раз в месяц, при наличии лейкоцитурии – посев мочи на флору с определением чувствительности к антибиотикам.
- Следить за стулом, не допускать появления запоров. Приучать опорожняться в одно и то же время – утром.
- Уродинамическое исследование и консультация нейроурологом через 6 мес.
В заключение хотим отметить, что пациентка с агенезией крестца требует частого уродинамического контроля для своевременной коррекции терапии. При таком условии на первый взгляд у малоперспективного пациента в плане восстановления тазовых функций представляется возможным улучшать качество его жизни, корригировать функцию нижних мочевыводящих путей и поддерживать пиелонефрит в ремиссии. После стабилизации накопительной функции мочевого пузыря (увеличение объема и снижение внутрипузырного давления) основной задачей является коррекция фазы опорожнения, уменьшение функциональной инфравезикальной обструкции.


