Оказание высококвалифицированной медицинской помощи на современном уровне не представляется возможным без медико-генетического обеспечения. От выяснения генетической основы того или иного заболевания во многом зависят успех его лечения и профилактика. По данным ВОЗ, более 90% всей патологии человека составляют болезни с наследственной предрасположенностью, за исключением моногенных и хромосомных болезней, имеющих исключительно генетическую природу возникновения. Клинические реакции инфекционной патологии тоже во многом зависят от генотипа. Расшифровка структуры генома человека открыла путь к пониманию молекулярных основ болезней, разработке принципиально новых стратегических подходов к их диагностике и лечению.
Мочекаменная болезнь (МКБ) занимает одно из ведущих мест в структуре урологических заболеваний и представляет одну из актуальных проблем современной урологии в связи с высокой распространенностью и склонностью к рецидивированию [1, 2]. Уролитиаз является третьим по встречаемости урологическим заболеванием, частота которого составляет 15–25%. Распространенность нефролитиаза среди населения России составляет: у детей – 19–20 случаев, у подростков – 80–82, у взрослых – 450–460 на 100 тыс. населения. Примерно в 65–70% случаев болезнь диагностируют у лиц в возрасте 20–25 лет, т.е. в наиболее трудоспособном периоде жизни [3, 4]. В подавляющем большинстве случаев в 80% причиной МКБ являются кальциевые конкременты (85–90% – оксалатно-кальциевые, 1–10% – фосфатно-кальциевые, 5% – оксалат и фосфат кальция в сочетании с мочевой кислотой). На мочекислые камни приходится 5–10%, на струвитные – 5–15%, на цистиновые – 1–3% [5, 6].Несмотря на внедрение современных методов лечения (дистанционная литотрипсия, чрескожная нефролитотрипсия), частота рецидивов остается на довольно высоком уровне и достигает 38,4–50,0% [7, 8]. Увеличение распространенности заболевания указывает на необходимость поиска факторов риска камнеобразования и в связи с этим – более пристального рассмотрения вопроса о роли наследственности и генетических критериев диагностики. К настоящему времени достаточно убедительно доказана тесная взаимосвязь генетических нарушений с клиническими проявлениями практически всех заболеваний человека, в том числе МКБ. Основную долю случаев МКБ, несомненно, следует отнести к мультифакториальной патологии, т.е. болезням с наследственным предрасположением, и лишь несколько десятков заболеваний (по данным каталога В. А. Маккьюсика), сопровождающихся уролитиазом, имеют моногенную природу наследования. И хотя точное число моногенных наследственных синдромов, сопровождающихся уролитиазом, назвать достаточно сложно и они относятся к редким наследственным заболеваниям, многие из них хорошо известны практикующим врачам-урологам (болезнь Дента, синдром Барттера, синдром Леша–Нихана и др.). Как уже отмечалось, гиперкальциурия, гипероксалурия считаются наиболее важными факторами риска МКБ. Из всех нарушений состава мочи чаще всего при МКБ отмечается гиперкальциурия, которая наблюдается у 40–50% больных. В табл. 1 представлены примеры моногенных синдромов МКБ, связанных с преобладанием кальциевых камней.
Как уже отмечалось, моногенные формы МКБ, сопровождающиеся гиперкальциурией, в основном являются редкими заболеваниями и проявляются сразу после рождения или в первые годы жизни ребенка. Не вызывает сомнений важность ранней и точной диагностики патологии с использованием молекулярно-генетических методов в связи с необходимостью своевременного назначения соответствующей терапии и профилактики заболевания у родственников пациента. В настоящее время молекулярно-генетическая диагностика уже успешно применяется для диагностики наследственных синдромов, связанных с образованием кальциевых камней, которые встречаются в клинической практике врача-уролога.
Болезнь Дента (Х-сцепленный рецессивный нефролитиаз) является аллельным вариантом Х-сцепленного тубулярного почечного нарушения, характеризующегося протеинурией, гиперкальциурией, нефрокальцинозом, нефролитиазом и почечной недостаточностью. Почечные депозиты состоят из кальция фосфата и кальция оксалата. Болезнь Дента обусловлена мутациями в гене хлоридного канала 5 (CLCN5), расположенном на хромосоме Xp11.22. Определены следующие мутации гена CLCN5: p.Trp279Ter-мутация привела к потере 469 аминокислот из D6-области до С-конца; p.Arg648Ter-мутация привела к потере 100 аминокислот с С-конце белка, удаление домена D13; p.Leu200Arg-мутация нарушила распределение заряда в домене D3 белка; p.Ser520Pro-мутация разрушила спираль в D11 [9]. Описан аномально большой экзон гена 11 CLCN5 [10, 11].
Гипофосфатемический нефролитиаз; остеопороз. Заболевание наследуется по аутосомно-доминантному типу. D. Prie и соавт. обнаружили две мутации гена натрий-фосфорного транспортера тип 2 SLC34A1: p.Аla48Phe и p.Val147Met, ген картирован на хромосоме 5q35.3. Авторы также описали 2 нуклеотидные замены в смежных положениях SLC34A1 гена: c.223G>T и c.224C>T. Мутантный белок привел к снижению обмена фосфатов. Наряду с потерей фосфора нарушается регуляция синтеза паратгормона и 1,25 (ОН)-витамина D, что может приводить к развитию гиперпаратиреоза, гиперкальциурии с формированием нефрокальциноза. Заболевание обычно манифестирует на 1–2-м годах жизни (иногда в более позднем возрасте – 5–6 лет) [12].
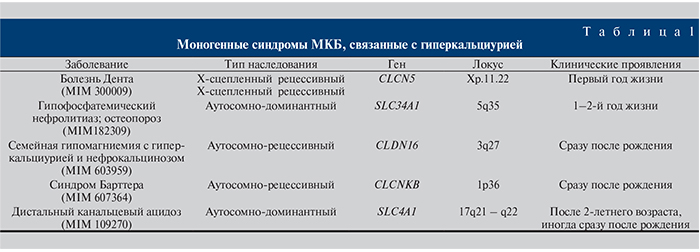
Семейная гипомагниемия с гиперкальциурией и нефрокальцинозом. Аутосомно-рецессивное заболевание обусловлено мутациями гена CLDN16 (PCLN1). Кодируемый геном белок локализуется в плотных контактах клеток петли Генле (толстой части ее восходящего отдела). Выявлен ряд мутаций гена CLDN16: p.Tyr277Ter, p.Thr303Arg, p.Lys275Ter, p.Leu151Pro, p.Leu151Trp, p.Gly191Arg, p.Leu151Phe, p.Gly198Asp, p.Met71Arg, p.Leu167Pro, p.Phe232Cys, p.Gly233Asp, p.Ser235Phe [13, 14]. S. Weber и соавт. проанализировали ген PCLN1 в 8 семьях и определили 8 различных мутаций. У гомозиготных по данным мутациям пациентов развивается заболевание, при котором больные теряют с мочой большое количество магния и кальция и у них возникают вторичная гипомагниемия и нефрокальциноз. Уровень кальция крови остается нормальным. Мутации PCLN1 приводят к массивной потере магния почками вследствие нарушения его транспорта. В последующем у больных развиваются нефрокальциноз и почечная недостаточность [15].
Синдром Барттера. Генетически обусловленная тубулопатия, которая наследуется по аутосомно-рецессивному типу. Причиной синдрома Барттера считают нарушение функции почечных канальцев, проявляющееся снижением реабсорбции ионов Cl- (и соответственно Na+)-клетками восходящего отдела петли Генле. Патогенез синдрома Барттера до конца не выяснен. Синдром может быть разделен на различные подтипы в зависимости от мутировавшего гена (NKCC2, ROMK, CLCNKB, BSND, CASR, SLC12A3). Классический синдром Барттера вызывается мутацией гена CLCNKB (локус 1p36) и проявляется выраженными нарушениями электролитного обмена (гипокалиемией), кислотно-щелочного равновесия (метаболическим алкалозом), гиповолемией, компенсаторной гиперплазией юкстагломеру-лярного аппарата почек и вторичным гиперальдостеронизмом. D. Simon и соавт. обнаружили, что дети с синдромом Барттера типа 3 были гомозиготными по одинаковой мутации – замене лейцина на пролин в кодоне 124-го гена CLCNKB [16].
Дистальный канальцевый ацидоз. Первичная тубулопатия наследуется по аутосомно-доминантному типу, обусловлена мутацией в гене SLC4A1. L. Bruce при изучении дистального канальцевого ацидоза описал часто встречающиеся мутации гена SLC4A1: p.Аrg589Cys, p.Ser613Phe, p.Arg589His, p.Arg589Ser [17]. Синдром проявляется метаболическим ацидозом, развивающимся вследствие нарушения подкисления мочи почками в отсутствие выраженного снижения функции клубочкового аппарата (нарушение секреции ионов водорода в дистальных или реабсорбции бикарбонатов в проксимальных отделах канальцев почек, что приводит к хроническому метаболическому ацидозу, гипокалиемии и нефрокальцинозу).
Моногенные синдромы, сопровождающиеся другими видами камнеобразования, встречаются еще реже, чем заболевания с кальциевыми конкрементами, однако они могут манифестировать в разное время, а значит, встречаться в разных возрастных группах урологических пациентов (табл. 2).
Диагностика этих заболеваний может представлять определенные сложности в связи с поздним проявлением и наличием у пациентов других хронических заболеваний. В таких ситуациях молекулярно-генетические исследования особенно важны, поскольку позволяют определять наличие мутаций в генах, ответственных за проявление определенной формы МКБ наследственной этиологии.
Синдром Леша–Нихана. Наследственное заболевание, вызванное дефектом гена гипоксантингуанинфосфорибозилтрансферазы (HPRT), расположенном на длинном плече Х-хромосомы. Ген участвует в обмене пуринов (аденина и гуанина) и кодирует гипо-ксантингуанинфосфорибозилтрансферазу, которая катализирует превращение гипоксантина в инозин монофосфат и гуанина в гуанозин монофосфат [17]. Ферментативный дефект приводит к нарушению пуринового обмена и повышенной продукции мочевой кислоты. У детей с данной патологией в раннем возрасте появляются тофусы, уратные камни в мочевыводящих путях и серьезные неврологические отклонения, сопровождающиеся нарушением речи, церебральными параличами, снижением интеллекта, склонностью к нанесению себе увечий. На сегодняшний день зарегистрировано более 600 мутаций в гене HPRT. К часто встречающимся мутациям можно отнести аминокислотные замены в 50-м положении Arg-Gly, в 109-м положении Ser-Leu, в 193-м положении Asp-Asn, в 103-м положении Ser-Arg [18].
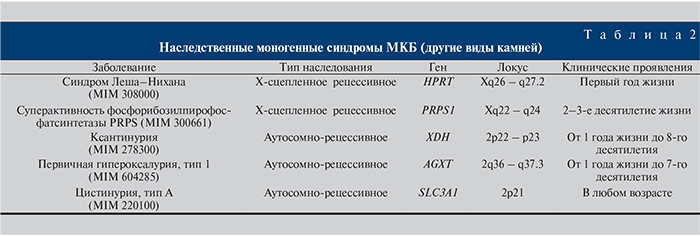
Суперактивность фосфорибозилпирофосфатсинтетазы (PRPS1). Х-сцепленный рецессивный врожденный дефект обмена веществ, при котором увеличение активности фермента ассоциировано с гиперурикемией и подагрой. У больных встречаются аномалии развития нервной системы, в частности нейросенсорная тугоухость, умственная отсталость, задержка развития опорно-двигательного аппарата. Описаны мутации в гене PRPS1 (p.Asn113Ser, p.Asp51His, p.Leu128Ile и p.Ala189Val), которые приводили к данной патологии. Патогенез оставался идентичным для всех видов мутаций [19, 20].
Ксантинурия, тип 1. Редкий дефект метаболизма пуринов, который развивается в результате мутаций в гене XDH и характеризуется экскрецией большого количества ксантина в моче со склонностью к образованию камней ксантина. P. Xu и соавт. описали ксантиндегидрогеназу как фермент, принадлежащий к группе молибденсодержащей гидроксилазы, вовлеченной в окислительный метаболизм пуринов. Классическим признаком заболевания является образование камней ксантина. Почечная недостаточность – одно из самых грозных осложнений для этой категории больных нефролитиазом. Среди других клинических проявлений у пациентов были отмечены язвенная болезнь двенадцатиперстной кишки, миопатия и атрофия мышц. Обнаружена молекулярная причина дефицита фермента и установлено наличие замены С-Т в нуклеотиде 682, что вызвало нонсенс-мутацию p.Arg228Ter и c.445C>T в экзоне гена XDH [21, 22].
Первичная гипероксалурия, тип 1. Заболевание развивается вследствие мутаций в гене аланинглиоксилат аминотрансферазы (AGXT), кодирующем пероксисомный фермент (ALT), локализующийся в печени. Снижение активности фермента ALT, катализирующего превращение глиоксилата в глицин, приводит к образованию из глиоксилата оксалатов, которые затем образуют нерастворимые соли кальция, накапливающиеся в почках и других органах и тканях. Первые симптомы патологии связаны с камнями в почках. Почечная колика или бессимптомная гематурия нарушают функции почек, что приводит к задержке роста и уремии. На сегодняшний день идентифицировано 146 мутаций гена AGXT. Наиболее часто встречалась мутация с.630 G > A, реже – c.588 G > A [23–25].
Цистинурия – наследственное заболевание, характеризующееся нарушением транспорта ряда аминокислот (цистина, лизина, аргинина, орнитина) в эпителиальных клетках почечных канальцев и кишечника и формированием цистиновых камней. Мутации гена SLC3A1 обусловливают бездействие мембранных транспортных систем, в результате этого реабсорбция цистина и (или) диаминомонокарбоновых кислот прекращается. При этом клиренс цистина может быть равным клиренсу инулина – эталона клубочковой почечной фильтрации (в среднем 127 мл/мин) или превышать его. Цистин плохо растворим в воде (концентрация насыщенного раствора цистина при рН, равном 7,0, составляет не более 400 мг/л). Превышение этого порога ведет к выпадению кристаллов цистина в осадок и образованию конкрементов. Выведение цистина с мочой у больных достигает 250 мг на 1 г креатинина в сутки и более (в норме 75–125 мг на 1 г креатинина в сутки). Определено 6 миссенс-мутаций в гене SLC3A1. Наиболее частые мутации в гене SLC3A1: CCA-CAAPro-Gln, TAC-AACTyr-Asn, CGG-CAG Arg-Gln, GAA-AAAGlu-Lys, ACA-GCAThr-Ala, ACG-ATGThr-Met, CGC-TGCArg-Cys [26, 27].
На сегодняшний день для каждого идентифицированного гена, мутации которого приводят к наследственному моногенному заболеванию, разработаны эффективные методы молекулярно-генетической диагностики, как правило, направленные на генотипирование наиболее частых мутаций этого гена. Реже для этих целей используют непрямой метод диагностики с помощью молекулярных маркеров. Выявление мутаций, связанных с моногенными болезнями, используют в целях диагностики этих заболеваний при наличии соответствующих клинико-фенотипических признаков. Кроме того, с помощью молекулярно-генетических методов можно оценить риск этих заболеваний для потомства в случае отягощенной наследственности у родителей. Таким образом, применение молекулярно-генетических методов при моногенной патологии позволяет идентифицировать весь спектр геномных нарушений, тем самым точно установить причину заболевания и предсказать возможность ее появления в потомстве.
Что касается мультифакториальных заболеваний, то расшифровка их этиопатогенеза значительно затруднена, так как в процесс развития конкретной формы заболевания вовлечены десятки генов, что значительно усложняет их диагностику. Исследования генома человека сделали реальной раннюю досимптоматическую диагностику многих мультифакториальных заболеваний, включая МКБ. Практически это достигается путем тестирования генов предрасположенности.
Гены предрасположенности можно определить как гены, наследственные варианты (полиморфизмы) которых совместимы с жизнью, но при воздействии неблагоприятных факторов среды могут стать причиной заболеваний. Уже известны сочетания таких генов (генные сети) для многих мультифакториальных заболеваний. В настоящее время появилось много отечественных и зарубежных исследований, посвященных поиску полиморфизмов кандидатных генов, участвующих в формировании уролитиаза, что может значительно облегчить раннюю диагностику патологии. В табл. 3 представлены исследованные в популяциях разных стран полиморфизмы ряда генов, ассоциированных с МКБ.
Ген VDR кодирует рецептор, связывающий витамин D3 (кальцитриол), регулирует активность генов минерального обмена и секрецию паращитовидного гормона, контролируя таким образом гомеостаз кальция и фосфора. T. Arcidiacono и соавт. исследовали ассоциацию гена VDR (12q12–14) с нефролитиазом в итальянской популяции. В результате были опре-делены три полиморфизма гена VDR на 3'-концевой области (rs121909790, rs121909791, rs121909800) с использованием BSMI (B/B аллели), ApaI (А / а), TaqI (т/T) рестрикционных ферментов. Первые два полиморфизма находились в пределах интрона 8, третий был в экзоне 9. Установлено, что аллели, снижающие экспрессию или активность VDR (В и Т), были связаны с риском камнеобразования. К тому же гомозиготные пациенты характеризовались более ранним возрастом манифестации МКБ и более быстрым ростом конкрементов [28]. В российской популяции поиск полиморфных вариантов кандидатных генов осуществляли О. И. Аполихин и соавт. [29], которые не обнаружили ассоциации уролитиаза с полиморфизмом гена рецептора витамина D (VDR, rs1540339).
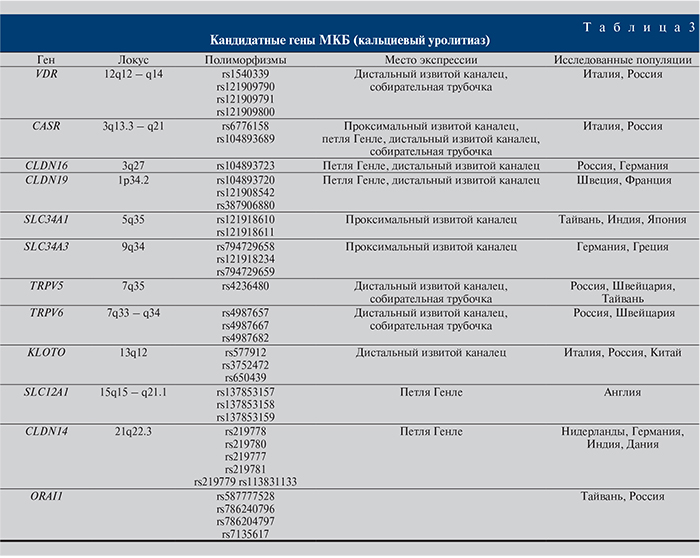
CASR является рецептором плазматической мембраны, который экспрессируется в паращитовидных железах и клетках, выстилающих канальцы почек. CASR реагирует на минимальные изменения концентрации кальция и передает эту информацию через внутриклеточные сигнальные пути, которые изменяют секрецию паратиреоидного гормона. Клеточный дефект выражается в ингибировании реабсорбции кальция в восходящей ветви петли Генле. G.Vezzoli и соавт. изучали полиморфизмы регуляторной области CASR гена (локус 3q21.1) с целью выявить взаимосвязь с идиопатическим кальциевым камнеобразованием в итальянской популяции. В результате установлена связь SNP (rs6776158) регуляторной области гена CASR, расположенного в пределах промотора 1, с образованием кальциевых камней [30]. В более поздних исследованиях обнаружены однонуклеотидные полиморфизмы (SNP) в экзоне 7 и регуляторной области гена CASR, связанные с развитием нефролитиаза. В работе показано, что рецессивный аллель Arg990-GlySNP (rs1042636, A>G), расположенный в экзоне 7, ассоциирован с развитием идиопатического нефролитиаза [31]. Анализ взаимосвязи полиморфизмов гена CASR с развитием камней почек в российской популяции не выявил статистически значимых данных [32].
CLDN16 – трансмембранный белок, локализующийся в плотных контактах клеток петли Генле (толстой части ее восходящего отдела). Нарушение функции белка приводит к потере с мочой больших количеств магния и кальция, вследствие чего возникают вторичная гипомагниемия и нефрокальциноз. Уровень кальция крови остается нормальным [33, 34]. D. Si-mon и соавт. еще в 1999 г. идентифицировали 10 различных мутаций в гене PCLN1, в том числе мутации сайта сплайсинга и миссенс-мутации (13). В последующем у больных развиваются нефрокальциноз и почечная недостаточность. M. Konrad и соавт. проанализировали 32 пациента с синдромом семейной гипомагниемии и нефрокальцинозом и в 7 из 13 мутантных аллелей обнаружили замену Leu151 на Phe, Leu151 на Pro [35].
Исследование в российской популяции среди детей [36] показало отсутствие ассоциации полиморфного локуса rs104893723 Gly198Asp гена CLDN16 с риском развития нефролитиаза и нефрокальциноза у детей с гиперкальциурией.
SLC34A1 – транспортный белок, который осуществляет перенос цистина через щеточную каемку эпителиальных клеток почечных канальцев. В результате мутаций нарушается процесс обратной резорбции и экскреции цистина через мембрану клеток канальцев, в просвет которых он попадает из плазмы крови после фильтрации через гломерулярные мембраны. Поскольку цистин слаборастворим, у больных цистинурией образуются цистиновые камни.
В японской популяции исследование полиморфизмов проводили T. Iwaki и соавт., которые установили, что полиморфизмы гена SLC34A1 rs121918610 и rs121918611 в экзоне 13 приводят к развитию гипофосфатемии, гиперкальциемии, повышению уровня щелочной фосфатазы, МКБ и гидронефрозу [37].
CLDN19 – трансмембранный белок, который контролирует транспорт ионов и молекул через эпителиальный слой, а также транспорт белков и липидов между апикальной и базолатеральной частями эпителиальных клеток [38]. Ген CLDN19 экспрессируется в нескольких органах, но более всего в почках и глазах. Нарушение функционирования белка приводит к изменению транспорта ионов в петле Генле. В результате развивается семейная гипомагниемия с гиперкальциурией и нефрокальциноз с офтальмологическими проявлениями. M. Konrad и соавт. охарактеризовали несколько шведских семей, имеющих тяжелую гипомагниемию с нефрокальцинозом и прогрессирующей почечной недостаточностью. Фенотип был практически не отличим от такового у больных другой формой семейной гипомагниемии, вызванной мутацией в гене CLDN16, хотя у этих пациентов не обнаружили никаких мутаций в гене CLDN16. CLDN19 был признан авторами наиболее перспективным кандидатным геномом при этом заболевании [39]. S. Faguer и соавт., изучая несколько французских семей с аналогичными клиническими проявлениями, описали несколько полиморфизмов гена СLDN19: rs104893720, rs121908542 и rs387906880 [40].
Ген SLC34A расположен в локусе 5q35.3, кодирует перенос фосфата NPT2a, ответственного за 85% реабсорбции фосфата в проксимальных канальцах [41]. Полиморфизмы гена SLC34A1 вызывают потерю функции NPT2a и приводят к развитию у больных гипофосфатемической гиперкальциурии. D. Dasgupta и соавт. описали несколько больных в немецкой популяции с гипофосфатемической гиперкальциурией и полиморфизмом гена SLC34A3: rs794729658, rs121918234 и rs794729659 [42].
TRPV5 и TRPV6 – гены семейства кальциевых каналов, имеют высокую степень гомологии и обладают сходными функциональными характеристиками. Эти каналы обеспечивают строго дозированное поступление кальция и участвуют в активной реабсорбции кальция в эпителиальных клетках почек, тонком кишечнике и плаценте [36].
M. Suzuki и соавт. обнаружили, что полиморфизмы гена TRPV6 C157R (rs4987657), M378V (rs4987667), M681T (rs4987682) определялись как Arg157, Val378 и Thr681 (RVT) и присутствовали у больных МКБ [43]. Исследование, проведенное в российской популяции среди детей [36], показало, что с риском развития нефролитиаза и нефрокальциноза у детей с гиперкальциурией ассоциированы полиморфизмы гена TRPV6. Предрасполагающими являлись гомозиготный генотип по T-аллелю (ТТ полиморфизма rs4987682 C2042TThr681Met гена TRPV6) и генотип AA полиморфизма rs4987667 G1132AVal378Met гена TRPV6. При обследовании 365 больных в популяции Тайваня обнаружена значимая связь между полиморфизмом гена TRPV5, rs4236480 и формированием множественных камней почек [44].
KLOTHO – трансмембранный белок 1-го типа с глюкуронидазной активностью, экспрессируется в тканях, ответственных за гомеостаз кальция, таких как почки и паращитовидные железы. D. Telci и соавт. были исследованы полиморфизмы G395A в области промотора, F252V в экзоне 2 и C1818T в экзоне 4; установлено, что GG-генотип G395A KLOTHO ассоциирован примерно с 2-кратным увеличением риска камнеобразования по сравнению с гомозиготным генотипом АА и гетерозиготным генотипом GA [45]. В российской популяции в исследовании зависимости между развитием МКБ и геномом KLOTHO обнаружено не было [3]. В ходе изучения полиморфизмов гена KLOTHO (rs3752472 в экзоне 3, rs650439 в интроне 4 и rs577912 в интроне 1), проведенного в китайской популяции, установлено, что только rs3752472 имеет достоверную связь с развитием МКБ [46].
SLC12A1 – семейство генов интегральных мембранных белков, которые опосредуют перенос Na+, K+ и Cl- через мембрану в восходящей ветви петли Генле. D. Simon и соавт. [47] в 1996 г. определили 6 мутаций в гене SLC12A1 в виде сдвига рамки считывания или аминокислотных замен. J. Halbritter и соавт. при исследовании популяции англичан не обнаружили взаимосвязи между полиморфизмами SLC12A1 (rs137853157, rs137853158, rs137853159) и развитием уролитиаза [48, 49].
CLDN14 является членом семейства генов мембранных белков, которые регулируют парацеллюлярный перенос ионов. В 2009 г. были опубликованы результаты первого исследования ассоциации гена CLDN14 с развитием уролитиаза, согласно которым МКБ ассоциировалась с двумя вариантами (R81R и T229T) гена CLDN14. Примечательно, что эти варианты также выявили связь с биохимическими показателями, имеющими отношение к обмену кальция [50]. H. Toka описал SNP гена rs113831133, при котором происходило увеличение экскреции Са2+ [51]. G. Thorleifsson и соавт. в европейской популяции определили четыре полиморфизма (rs219778, rs219779, rs219780 и rs219781) в гене CLDN14, связанных с образованием мочевых камней, что согласовывается с результатами работы [52] из Индии, в которой обнаружено три SNP (rs219777, rs219778 и rs219780), среди них два (rs219778 и rs219780) были достоверно связаны с МКБ [52].
ORAI1 – ген, кодирующий мембранный белок плазмы, необходимый для трансмембранного обмена кальция. Y. Chou и соавт. [53] изучали полиморфизмы гена ORAI1 и показали, что полиморфизмы (rs587777528, rs786240796 и rs786204797) могут являться генетическими факторами риска возникновения и рецидива МКБ. Исследование [29] свидетельствует о том, что на формирование множественных камней при уролитиазе могут влиять генетические факторы, в частности полиморфные варианты гена модулятора активатора высвобождения кальция 1 ORAI1, rs7135617.
В данной работе мы обобщили данные по исследованиям далеко не всех генов, влияющих на формирование мочевых камней. Несомненно, исследования по выявлению генных полиморфизмов МКБ продолжаются учеными разных стран ввиду актуальности мочекаменной болезни. Развитие технологий анализа ДНК привело к разработке методов секвенирования нового поколения (Next Generation Sequencing – NGS) и других постгеномных технологий, в том числе полногеномного исследования ассоциаций (Genome-Wide Association Study – GWAS), что позволяет выделять редкие генные варианты, вносящие значительный вклад в риск развития многих заболеваний человека, включая МКБ. Современные молекулярно-генетические технологии позволяют оценивать наследственную предрасположенность к заболеваниям. Основным препятствием к использованию этих технологий в клинической практике являлась их высокая стоимость для пациентов. Однако благодаря постоянно снижающейся стоимости, высокой производительности, а также широким возможно-стям мультиплексирования по образцам и тестируемым регионам технология NGS в ближайшем будущем должна прийти на смену целому спектру методов ДНК-диагностики. Трактовка полученных результатов таких исследований требует привлечения квалифицированных и опытных специалистов, разбирающихся как в генетических аспектах, так и в особенностях группы нарушений, для которой выполняется генетическое тестирование. В связи с этим совершенно очевидно, что как для выяснения этиологических факторов в случае моногенных форм МКБ, так и для определения генетической компоненты в случае мультифакториальной природы заболевания необходимо проведение медико-генетического консультирования.
Постоянное развитие и совершенствование молекулярно-генетических технологий позволяют понять, что причиной все большего числа болезней, которые раньше связывались только с неправильным питанием, напряженным ритмом жизни, возрастом и т.п., являются и нарушения в функционировании различных генов. Выявить такие нарушения, составить объективный прогноз и выбрать оптимальную схему лечения, а в ряде случаев и предотвратить заболевание – задача превентивной и персонализированной медицины будущего.
